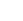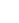专业科学仪器及设备制造商

DOI:10.1039/D6EE01390B
全文速览
析氢反应(HER)在碱性介质中的动力学比酸性介质中慢约两个数量级,这一长期未解的“动力学pH效应”严重制约了阴离子交换膜水电解器(AEMWE)的效率。本研究通过晶界工程在PtIr纳米合金中引入高密度位错和拉伸应变,调节表面亲氧性,从而重构电极/电解液界面处氢键网络的连接性,而非仅仅改变反应热力学能垒。结合原位光谱和理论计算,研究揭示了应变诱导的富电子表面增强OH吸附,进而增加界面间隙区水分子的比例(从12.9%增至29.0%),强化氢键网络连通性,促进水分子和质子传递。最优催化剂GB‑PtIr@CNT在碱性和酸性介质中的HER活性几乎无差异,Tafel斜率均为~25.5 mV dec⁻¹。在AEMWE器件中,超低PGM载量(90 μg cm⁻²)下达到2 A cm⁻²仅需1.84 V,并稳定运行1000 h。该工作为打破pH依赖动力学提供了基于界面水重构的实用策略。
背景介绍
氢经济的关键技术之一是高效水电解制氢。AEMWE因其可使用非贵金属析氧催化剂而受到关注,但其阴极析氢反应在碱性环境中动力学极慢。以Pt为例,从酸性到碱性介质,反应速率下降约两个数量级,这种“动力学pH效应”根源至今未完全阐明。传统理论(如氢结合能理论、双功能机理、2B理论等)主要从热力学角度解释,但难以解释非能斯特型的pH依赖性。近年研究表明,界面双电层中的水分子氢键网络连接性对质子/水传递至关重要,而OH吸附可以调控该网络。然而,如何在不牺牲活性位点或引入额外物种的前提下精准调控界面微环境,仍具挑战。本研究利用晶界诱导的拉伸应变,在不改变催化剂本征热力学路径的前提下,仅通过调节界面水结构,成功打破了HER的pH效应。
本文亮点
(1)晶界工程创制高密度位错:通过PtIr合金纳米粒子构建高密度晶界(表面密度达872.6 µm⁻¹),引入6.8%拉伸应变,使d带中心上移,表面亲氧性增强,OH吸附能力提升。
(2)界面水氢键网络重构:原位ATR‑SEIRAS揭示应变催化剂使界面间隙区水分子比例从12.9%大幅增至29.0%,强化氢键网络连通性,促进水/质子传递,而非改变Volmer步骤能垒。
(3)打破pH效应:GB‑PtIr@CNT在碱性与酸性介质中HER性能几乎无差(η₁₀₀均为~45 mV,Tafel斜率均为~25.5 mV dec⁻¹),质量活性间隙因子仅1.02(Pt/C为4.65),实现近pH不依赖催化。
(4)器件级验证:AEMWE在1.84 V达2 A cm⁻²,1000 h稳定运行(降解率23.1 μV h⁻¹),结构保持完整。
图文解析
图1:催化剂合成与晶界结构表征
(a) 碳纳米管负载晶界结构PtIr纳米合金(GB‑PtIr@CNT)的设计示意图,x:y为原子比;(b, c) GB‑PtIr@CNT不同区域的HAADF‑STEM图像,红色虚线标出晶界;(d) (b)中A1区域和(c)中A2区域的FFT花样;(e) GB‑PtIr@CNT的EDS元素面分布图;(f) 对应(c)中区域的几何相位分析(GPA)应变分布图,色标表示面内应变强度;(g) (f)中感兴趣区域(ROI)从左到右的相对应变分布;(h) GB‑PtIr@CNT中五重和低重晶界的表面密度;(i) GB‑PtIr@CNT及对照催化剂的拉曼光谱;(j) GB‑PtIr@CNT及对照催化剂的C K边sXAS谱(TEY模式)。
HAADF‑STEM(图1b‑c)清晰显示高密度晶界(红虚线),FFT证实五重孪晶结构。EDS(图1e)显示Pt、Ir均匀分布。GPA(图1f‑g)定量给出平均拉伸应变为6.8%。晶界表面密度高达872.6 µm⁻¹(图1h),远超常规晶界富集催化剂(20‑100 µm⁻¹)。Raman(图1i)和sXAS(图1j)表明碳载体结构未变,排除了金属‑载体相互作用的干扰。
图2:电子结构与配位环境
(a) Pt 4f和Ir 4f高分辨XPS谱;(b) 价带谱;(c) 归一化Pt L₃边和(d) Ir L₃边XANES谱;(e) Pt L₃边EXAFS的傅里叶变换谱(R空间);(f‑h) Pt foil、GB‑PtIr@CNT和PtO₂的k²加权Pt L₃边EXAFS小波变换图;(i) GB‑PtIr@CNT及对照催化剂的Pt‑Pt/Ir键长与d带中心关系;(j) 拉伸应变调控亲氧性的d带中心示意图。
XPS图谱显示GB‑PtIr@CNT的Pt 4f和Ir 4f结合能均负移,表明电子富集。UPS(图2b)中d带中心上移至‑5.18 eV(c‑PtIr为‑5.61 eV),利于OH吸附。XANES(图2c‑d)表明合金形成。EXAFS拟合结果显示GB‑PtIr@CNT的Pt‑Pt/Ir键长最长(2.74 Å),且应变与d带中心正相关(图2i),证实拉伸应变使d带上移、亲氧性增强(图2j)。
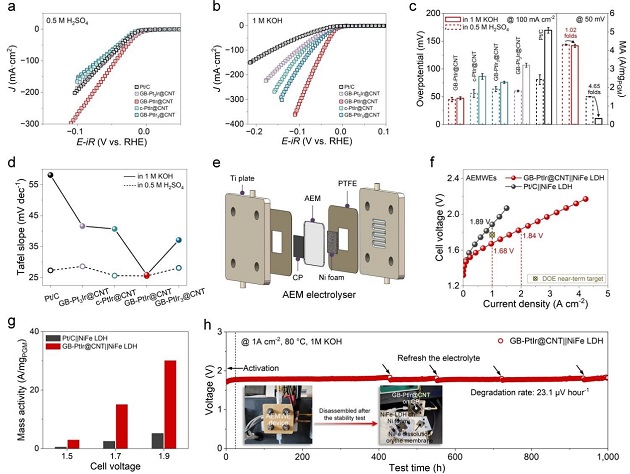
图3:电化学HER性能与AEMWE器件
(a)0.5 M H₂SO₄和(b) 1 M KOH中的HER极化曲线;(c) 各催化剂在η=100 mA cm⁻²时的过电位(左)及η=50 mV时的质量活性(右),并给出酸/碱质量活性间隙因子;(d) 塔菲尔斜率;(e) AEMWE膜电极示意图;(f) GB‑PtIr@CNT和Pt/C在1 M KOH、80 ℃下的AEMWE极化曲线;(g) GB‑PtIr@CNT和Pt/C的质量活性对比;(h) AEMWE耐久性测试,插图为运行前后器件照片及拆解后照片。
GB‑PtIr@CNT在碱性和酸性中的η₁₀₀分别为47.3和45.1 mV,几乎相同(图3c)。质量活性间隙因子仅1.02(Pt/C为4.65)。Tafel斜率在酸碱中均为~25.5 mV dec⁻¹,打破pH依赖。AEMWE达到2 A cm⁻²仅需1.84 V,质量活性远超Pt/C,1000 h稳定运行,晶界结构保持。
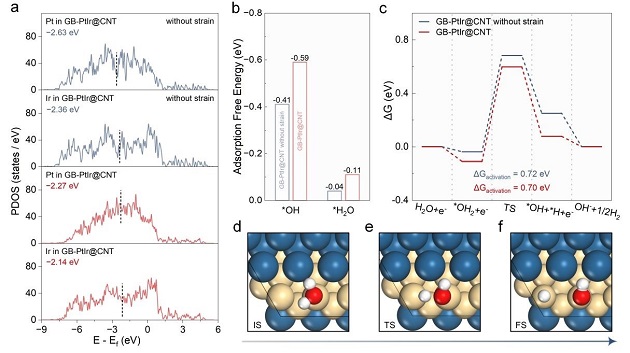
图4:应变对电子结构及反应能垒的影响
(a) 无应变(上)和拉伸应变(下)下GB‑PtIr@CNT中Pt和Ir的d轨道态密度;(b) 施加应变前后OH和H₂O吸附自由能变化;(c) 无应变和应变下的HER吉布斯自由能曲线;(d‑f) 水离解步骤的初始态、过渡态和最终态结构(蓝:Pt,黄:Ir,红:O,白:H)。
应变使Pt和Ir的d带中心分别上移0.36和0.22 eV,增强OH和H₂O吸附(图4b)。但水离解能垒仅从0.72降至0.70 eV,变化甚微,说明性能提升并非源于热力学能垒降低,而是界面水结构改变。
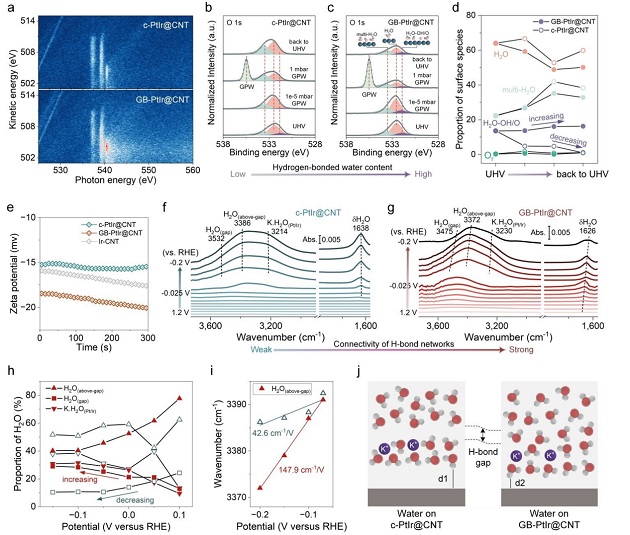
图5: 原位光谱揭示界面水重构机制
(a) 水气氛下彩色多维共振俄歇光谱(mRAS)。(b) c‑PtIr@CNT和(c) GB‑PtIr@CNT在不同水压下的APXPS O 1s谱;(d) 三种表面物种的比例变化(实心:GB‑PtIr@CNT;空心:c‑PtIr@CNT);(e) Zeta电位对比。(f) c‑PtIr@CNT和(g) GB‑PtIr@CNT在1 M KOH中不同电位下的原位ATR‑SEIRAS(O‑H伸缩振动);(h) 不同电位下界面水三种构型的比例(空心:c‑PtIr@CNT;实心:GB‑PtIr@CNT);(i) H₂O(above‑gap)的电位依赖峰位移;(j) 双电层中水分布示意图。
APXPS(图5b‑d)表明GB‑PtIr@CNT表面能稳定吸附更多氢键水。ATR‑SEIRAS(图5g‑h)显示,随电位负移,GB‑PtIr@CNT的间隙水比例从12.9%增至29.0%,而c‑PtIr从24.3%降至10.4%,证明应变催化剂强化了氢键网络连通性。SHINERS表明GB‑PtIr@CNT在HER条件下保持更高OH覆盖度,促进水网络重构。图5j总结:应变增强OH吸附→间隙水富集→氢键网络连通→加速质子/水传递→打破pH效应。
总结与展望
本研究通过高密度晶界工程在PtIr纳米合金中引入拉伸应变,系统揭示了HER动力学pH效应的来源并非传统热力学能垒,而是界面水氢键网络连接性。应变增强亲氧性,使OH吸附稳定,进而重构双电层中水的分布,增加间隙区水比例,强化氢键网络,从而加速水分子和质子转移,实现近pH不依赖的HER性能。最优催化剂在AEMWE器件中表现出创纪录的活性和稳定性。该工作为电催化界面微环境调控提供了全新思路:通过晶界应变而非外来添加剂来优化界面水结构。未来可拓展至其他pH敏感反应(如CO₂还原、氧还原),并探索非贵金属的晶界工程策略,推动绿色氢能技术发展。
通讯作者简介
叶逸凡,中国科学技术大学国家同步辐射实验室,特任研究员,博士生导师。2016年于中科大获得博士学位,长期从事同步辐射原位谱学仪器设备、表征技术和实验方法学的研发及应用拓展。在原子尺度上精准构高活性、高稳定性和高选择性的催化剂,利用多种同步辐射原位谱学手段和配套原位反应装置,实现催化剂表界面化学态、电子态的精确指认和解析,取得了一系列学术成果包括Science, Nat. Catal., Nat. Comm., J. Am. Chem. Soc., Angew. Chem. Int. Ed., 等。
本文使用的焦耳加热装置由合肥原位科技有限公司研发,感谢老师支持和认可!

焦耳加热装置
焦耳加热装置是一种新型快速热处理/合成的设备,该设备可使材料在极短(毫秒级/秒级)时间内达到极高的温度(1000~3000℃),升温速率最快可达到10000k/s;通过对材料的极速升温,可考察材料在极端环境、剧烈热震情况下的物性改变,可通过极速升降温制备纳米尺度颗粒,单原子催化剂,高熵合金等。目前广泛应用在电池材料、催化剂、碳材料、陶瓷材料、金属材料、塑料降解、生物质等领域。