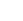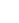专业科学仪器及设备制造商

DOI:10.1002/adma.202408906
全文速览
在CO2加氢反应中,通常认为氧空位主要对CO2吸附和活化步骤起着重要作用。然而,根据氧空位的结构与电子特性,以及CO2转化反应的连续性,氧空位对反应过程中一系列含氧中间体的作用可能长期被忽视和低估。本文以CO2光甲烷化为模型反应,利用富氧空位的Co3O4来研究氧空位与含氧中间体形成、转化的关系。原位漫反射傅立叶变换红外光谱(DRIFTS)分析和密度泛函理论(DFT)计算结果表明,甲酸盐是反应过程的关键中间体,其C–O键断裂是Co3O4表面甲烷化反应的限速步骤。而氧空位的存在可加速含甲酸盐在内的各含氧中间体的C–O键断裂及转化,进而实现较高的CH4生成速率(1108.1 mmol g−1 h−1)和选择性(93%)。这项研究为进一步认识CO2转化反应中氧空位的多重作用提供了一定的见解,为后续设计和开发高性能CO2转化催化剂提供了实验支撑。
背景介绍
人为排放的温室气体CO2浓度不断升高,造成了冰川融化,海平面上升等一系列环境问题,对自然环境构成了严重的威胁。光驱动的CO2加氢转化为高附加值的燃料和化学品,被认为是转化和利用温室气体CO2的有效策略。虽然氧空位被广泛认为是氧化物基催化剂的活性位点,但关于其在CO2加氢过程中如何发挥作用的认知有限,迫切需要对其综合作用进行深入的研究和揭示。为了解决这一问题,作者采用结构定制的Co3O4(富氧空位的介晶Co3O4,M-Co3O4)与商用Co3O4(C-Co3O4)作为对比,以CO2光甲烷化为模型反应,用于研究氧空位在含氧中间体形成和转化过程中扮演的角色。
本文亮点
1. 本工作构建了具有特定晶面暴露特征的富氧空位Co3O4(M-Co3O4)。
2. 原位漫反射傅立叶变换红外光谱(DRIFTS)分析和密度泛函理论(DFT)计算揭示了甲酸盐(HCOO*)是关键中间体,其C–O键断裂过程是Co3O4催化剂表面CO2光甲烷化反应的限速步骤。
3. M-Co3O4催化剂的氧空位加速了甲酸盐及其它含氧中间体的C–O键断裂,从而实现了优异的CH4生成速率(1108.1 mmol g–1 h–1)和高选择性(93%)。
图文解析
为了获得特定晶面暴露的Co3O4催化剂,作者采用水热和煅烧法构建了富含氧空位的介晶Co3O4(M-Co3O4,图1)。M-Co3O4呈现出薄片穿插状结构,并且在其矩形薄片中可以清晰观察到大量的孔隙(图1a–c,i–k)。通过透射电镜分析得到M-Co3O4的晶格间距为0.28 nm,这与Co3O4的{220}晶面族十分吻合(图1d)。从多孔矩形薄片的不同位置收集到的电子衍射斑点分布几乎相同,表明M-Co3O4多孔薄片表现出介晶的性质(图1e,f)。根据M-Co3O4多孔薄片的晶带轴[111]以及测得的(![]() 02)和(2
02)和(2![]() 0)面,可推断出M-Co3O4优先暴露(111)晶面。这种晶体结构配置可使M-Co3O4表面富集Co2+,并且更容易形成氧空位(图1g,h)。
0)面,可推断出M-Co3O4优先暴露(111)晶面。这种晶体结构配置可使M-Co3O4表面富集Co2+,并且更容易形成氧空位(图1g,h)。

图1.富氧空位M-Co3O4催化剂的结构表征
随后,作者采用多种表征技术(如图2所示),包括X射线光电子能谱(XPS)、电子顺磁共振(EPR)、正电子湮灭谱(PAS)和X射线吸收精细结构谱(XAFS)等,来研究催化剂的化学状态、电子结构、缺陷类型和配位环境,并藉此证实相比于商品Co3O4(C-Co3O4),结构定制的M-Co3O4含有大量的氧空位。
图2. 钴基催化剂的结构表征
图3 钴基催化剂的CO2加氢性能测试
为了深入揭示CO2加氢性能与催化剂表面氧空位之间的关系,作者在固定床反应器中使用CO2和H2的混气(CO2/H2,1/4),对M-Co3O4和C-Co3O4催化剂进行了催化性能测试,如图3所示。M-Co3O4催化剂展现出较高的CH4生成速率(1108.1 mmol g–1 h–1)及选择性(93%)(图3a)。这表明M-Co3O4在CO2加氢反应中活性优异,能够有效地促进CH4的生成。相比之下,对照样品也表现出较高的CH4生成速率(C-Co3O4为402.9 mmol g–1 h–1,CoO为217.1 mmol g–1 h–1,Co为282.2 mmol g–1 h–1),但均远低于M-Co3O4。为了进一步确认CH4产物的C元素来源,作者以13CO2为C源进行同位素标记实验,证实CH4是通过CO2加氢产生的(图3b)。此外,M-Co3O4催化剂在5000 mL g–1 h–1的高空速条件下可持续运行30小时,CH4选择性保持在96%以上,CO2转化率高达54.04%,TON(以Co摩尔数为基)达66.17,这表明M-Co3O4具备优异的CO2加氢活性和良好的稳定性(图3c)。
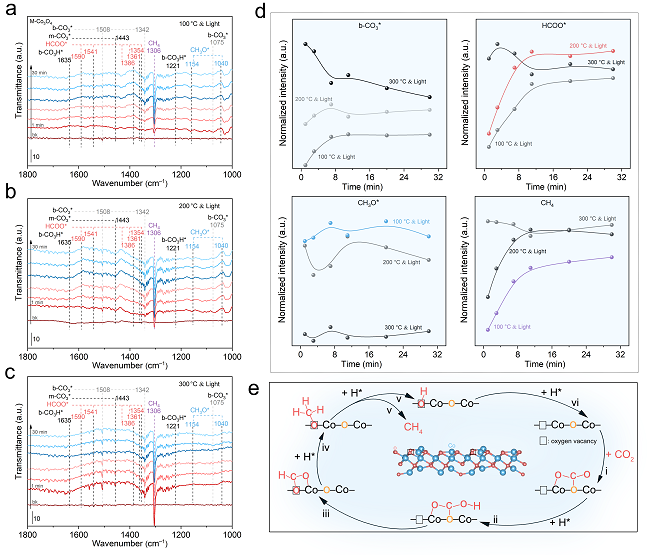
图4. CO2加氢反应的DRIFTS机理研究
为了探索CO2加氢过程的反应机理和反应路径,作者利用原位红外光谱技术监测了CO2光加氢过程中Co3O4催化剂表面吸附物种和反应中间体的演化。当M-Co3O4和C-Co3O4催化剂暴露于CO2/H2(1/4)混气时,可在其表面检测到CO3H*、CO3*、HCOO*和CH3O*物种(图4a–c)。图4d展示了M-Co3O4催化剂表面各含氧中间体的演变趋势。通过对上述各含氧中间体演变趋势进行分析可知CO2光甲烷化的反应路径为:CO2→CO3*/CO3H*→HCOO*→CH3O*→CH4。而HCOO*的演变趋势很大程度上影响了最终产物CH4的生成,故推测HCOO*可能是CO2光甲烷化反应的关键中间体,其C–O键断裂可能是限速步骤。
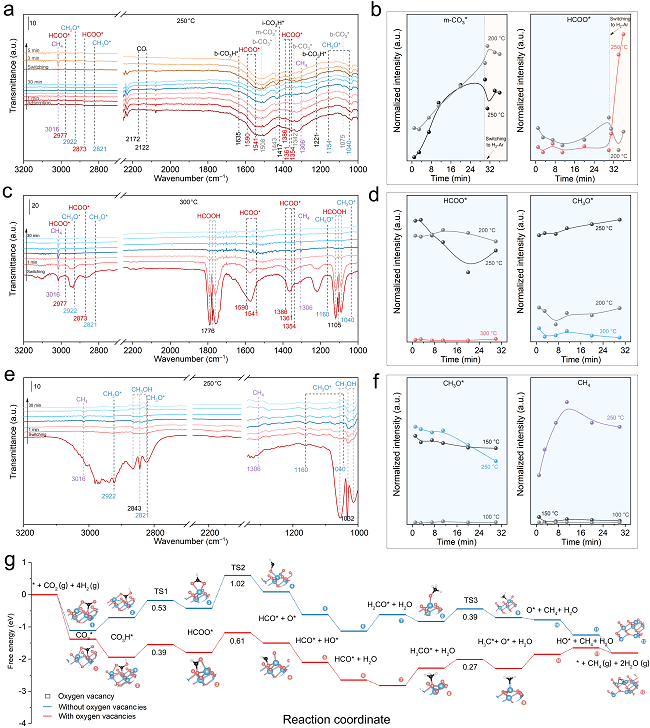
图5. M-Co3O4催化剂表面碳酸盐(CO3*)、甲酸盐(HCOO*)和甲氧基(CH3O*)物种的转化过程。
为了进一步阐明氧空位对各含氧中间体的作用,并证实HCOO*是CO2甲烷化反应的关键中间体,其C–O键的断裂是反应限速步骤,作者开展了一系列原位DRIFTS实验进行深入研究。具体来说,通过模拟反应中间体及反应氛围(CO2 + H2/Ar、HCOOH + H2/Ar 和CH3OH + H2/Ar),并监测目标中间体的演化行为,以期证实HCOO*物种的缓慢转化过程及氧空位在该过程中的独特作用。
图5a,b展示了M-Co3O4催化剂表面CO2吸附和转化的情况。在温度≤250°C时,b-CO3*物种立即形成并且保持基本恒定的浓度(图5a,b);相比之下,m-CO3*物种的红外峰面积(即浓度)随着CO2暴露时间的增长而逐渐增大。将上述吸附CO2后的催化剂切换至H2/Ar氛围后发现,当温度为200°C时,b-CO3*和m-CO3*物种的转化缓慢,而HCOO*及CH4均处于较低含量水平(图5b);而当温度达到250℃时,b-CO3*和m-CO3*物种含量逐渐衰减,与此同时HCOO*和CH4的红外吸收逐渐增强(图5a,b)。上述实验表明b-CO3*和m-CO3*物种的临界转化温度接近250℃,而在C-Co3O4表面也发现了类似的现象,尽管b-CO3*和m-CO3*物种的转化以及CH4产物的形成要慢得多。
为了研究甲酸盐(HCOO*)中间体的转化行为并阐明氧空位的相关效应,作者将甲酸引入DRIFT系统,并监测其在H2/Ar氛围下的相应变化(图5c,d)。在≤250°C时,气态HCOOH有微量消耗,HCOO*向CH3O*物种的转化可以忽略不计,此时没有检测到CH4产物(图5d);当温度提升至300°C时,HCOO*和CH3O*物种立即处于低浓度水平,而CH4产物显著增长(图5c,d)。这些实验结果表明,HCOO*物种的快速消耗和有效转化发生在大约300°C。而在同等条件下测试氧空位较少的C-Co3O4时,HCOO*的消耗和CH4的生成速度较慢,这表明氧空位在HCOO*中间体的C–O键断裂过程中发挥了促进作用。
针对CH3O*物种的转化行为,作者将甲醇引入DRIFT系统,监测了其在H2/Ar氛围下的转化过程(图5e,f)。在100℃和150℃时,尽管CH3OH和CH3O*物种的消耗速度不同,但并未观察到CH4的生成(图5f);相比之下,反应温度为250℃时可立即检测到气相CH4产物(图5e,f)。在C-Co3O4催化剂表面也得到了类似的实验结果,尽管其CH4生成速度比M-Co3O4慢。这些分析结果表明,CH3O*物种所需的转化温度约为250°C或更低。
结合原位红外实验分析和DFT计算结果,我们描绘出氧空位作用下CO2甲烷化反应的完整过程(图4e):i,游离CO2与M-Co3O4的晶格O结合形成CO3*;ii,活性H向CO3*转移并生成CO3H*;iii,CO3H*进一步加氢裂解C–O键生成HCOO*,其中一个O原子填补氧空位;iv,HCOO*中连接Co位的C–O键断裂,并逐渐氢化形成CH3O*,其O原子留在氧空位上;v,持续氢化导致CH3O*中C–O键断裂,释放CH4;vi,被O原子占据的氧空位通过H2的还原得到再生,从而完成催化循环。
总结展望
在这项研究中,作者采用富含氧空位的Co3O4(M-Co3O4)来深入探讨氧空位在CO2光甲烷化过程中的具体作用。研究揭示了甲酸盐(HCOO*)是反应过程的关键中间体,它的C–O键断裂是CO2光甲烷化过程的限速步骤。M-Co3O4的氧空位加速各含氧中间体(包括CO3*/CO3H*、HCOO*和CH3O*)C–O键的断裂,从而促进了其转化,尤其是转化较慢的甲酸盐关键中间体,因此实现了优异的CH4生成速率。这项研究深化了对CO2转化反应中氧空位关键作用的认识,为设计和优化高效CO2(光)加氢催化剂提供有价值的实验支撑。
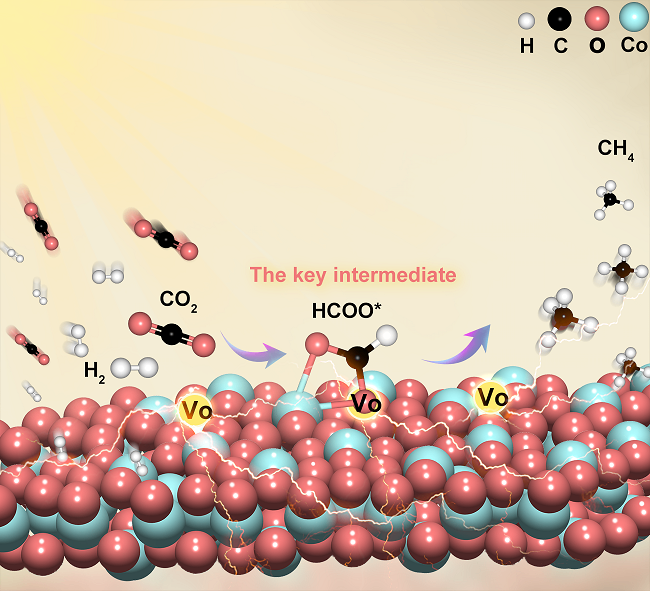
(TOC)
文章信息
Zhexing Lin, Zhengwei Yang, Jiajia Wang, Jun Wang, Huiting Huang, Jianyong Feng*, Huihui Yan, Minyue Zhao, Xinyi Liu, Wangxi Liu, Zhaosheng Li*, Zhigang Zou, Unlocking the Potential of Oxide-based Catalysts for CO2 Photo-hydrogenation: Oxygen Vacancies Promoted C–O Bond Cleavage in Key Intermediates, Adv. Mater. 2025, 2408906, https://doi.org/10.1002/adma.202408906
致谢
南京大学博士生林哲荇、博士生杨争伟、河海大学副教授王家佳为该项成果共同第一作者。南京大学李朝升教授、冯建勇副教授为该论文通讯作者。该工作得到邹志刚院士支持与指导。感谢王骏博士、黄辉庭博士、博士生闫会会、博士生赵敏跃、硕士生刘欣仪、博士生刘望喜在数据分析上提供的帮助。该工作受到国家自然科学基金(国家杰出青年科学基金)、国家重点研发计划等项目支持,并得到固体微结构物理国家重点实验室等平台的大力支持。
本文使用的原位反应池由合肥原位科技有限公司研发,感谢老师支持和认可!